Deepminds AlphaFold 2.0 sagt Proteinfaltungen so gut voraus, dass Wissenschaftler des CASP die KI als Lösung für das 50 Jahre alte Problem der Proteinfaltung bezeichnen. Das könnte den Beginn einer zweiten genetischen Revolution bedeuten: Deepmind tauscht das Go-Brett gegen das Spiel des Lebens.
In jeder Zelle existieren Tausende Proteine. Sie ermöglichen das Leben – vom Einzeller bis zum riesigen Säugetier. Nahezu jede Krankheit, darunter Krebs, Demenz und auch COVID-19, hängen direkt mit der Funktion der Proteine zusammen.
Die Funktion wird maßgeblich von der dreidimensionalen Struktur eines Proteins mitbestimmt, der sogenannten Faltung. Wer diese Faltung kennt, kann mehr über die Biologie des Lebens verstehen, Krankheiten besser diagnostizieren und heilen oder spezielle Proteine etwa für Nahrungsmittel, Energiegewinnung, Müllentsorgung oder Bekleidung finden.
Ein mächtiges Rätsel
Einfach gesagt: Wer das Geheimnis der Proteinfaltung entschlüsselt, findet Antworten auf viele grundlegende Fragen der Menschheit.
Seit knapp 50 Jahren suchen Forscher daher nach Wegen, die Mechanismen der Proteinfaltung aufzudecken. Der amerikanische Biochemiker Christian Anfinsen zeigte in der Frühphase der Protein-Forschung, dass sich die Struktur eines Proteins aus seiner Proteinsequenz (Aminosäuresequenz) theoretisch ableiten lässt und erhielt dafür 1972 den Nobelpreis für Chemie.
Diese direkte Ableitung hat in der Praxis einen großen Vorteil: Wissenschaftler müssen seltener auf aufwendige Methoden wie Kernspinresonanzspektroskopie, Röntgenstrukturanalyse oder Kryoelektronenmikroskopie zurückgreifen. Stattdessen können sie den Suchraum möglicher Strukturvarianten einschränken, so ihre experimentelle Forschung fokussieren und schneller Fortschritte erzielen.
Proteinfaltung lässt sich nicht mit roher Gewalt lösen
Doch es gibt ein Problem: Die Anzahl theoretisch möglichen Varianten, in die sich ein Protein falten kann, ist gigantisch. Der Molekularbiologie Cyrus Levinthal schätzte die Anzahl auf 10 hoch 300 Möglichkeiten und bemerkte, dass die Berechnung aller möglicher Konfigurationen mehr Zeit benötige, als das Universum existiere.
Zahlreiche Wissenschaftler wie Strukturbiologen, Bioinformatiker und „Computational Biologists“ versuchen seither das Problem der Vorhersage von Proteinfaltungen zu lösen.
Bekannt ist das im Jahr 2000 gestartete Folding@Home-Projekt, das die Rechenleistung privater Computer, Spielekonsolen und Smartphones bündelt, um Proteinfaltungen vorherzusagen. Im März durchbrach das Netzwerk als erstes die Exascale-Grenze und im April erreichte das Projekt eine kombinierte Rechenleistung von 2,43 Exaflops. Es ist damit der schnellste Supercomputer der Welt – aber noch immer nicht schnell genug.

CASP sucht seit 26 Jahren nach der besten Vorhersagemethode
Denn trotz der geballten Rechenkraft ist nur ein Bruchteil der in der Natur vorkommenden Proteinstrukturen bekannt. Als Vergleich: Dank der Durchbrüche der Genomforschung verzeichnet die Universal Protein Database (UniProt) knapp 180 Millionen Proteinsequenzen. Die Protein Data Bank (PDB) enthält dagegen jedoch nur rund 170.000 bekannte Proteinstrukturen.
1994 gründeten die Professoren John Moult und Krzysztof Fidelis daher „Critical Assessment of Protein Structure Prediction“ (CASP), eine alle zwei Jahre stattfindende Überprüfung aktueller Methoden für die Vorhersage der Proteinfaltung. Das Ziel ist, die besten Methoden für die Vorhersage von Proteinstrukturen zu finden und so zu fördern.
Bei dem Wettbewerb werden die Teilnehmer aufgefordert, Proteinstrukturen vorherzusagen, die zwar bereits von anderen Wissenschaftlern empirisch aufgedeckt, aber noch nicht veröffentlicht wurden. Sie sind nur den beteiligten Wissenschaftlern und CASP bekannt. Dadurch kann CASP die Vorhersagen überprüfen, ohne dass die Teilnehmer zuvor Zugriff auf die empirischen Daten hatten.
Bereits 2018 gelang Deepmind ein großer Erfolg
2018 nahm das erste Mal Deepminds Vorhersage-KI AlphaFold an dem Wettbewerb teil. Die damals seit zwei Jahren in Entwicklung befindliche KI belegte auf Anhieb den ersten Platz von 98 Teams.
Sie sagte bei 25 von 43 Proteinen die akkurateste Struktur vorher. Wichtig ist, die Dimension dieses Sieges zu verstehen: Das zweitplatzierte Team derselben Kategorie konnte die Proteinfaltung bei nur drei Proteinen vorhersagen.
Schon damals war also klar, dass Deepmind ein wissenschaftlicher Durchbruch gelingen könnte. Demis Hassabis, Mitgründer von Deepmind, bezeichnete den CASP-Triumph als “Schlüsselmoment”, bei dem seine Firma erstmals bewiesen habe, dass KI “fundamentale, sehr wichtige und reale wissenschaftliche Probleme lösen” könne.
Ein Beispiel für eine erfolgreiche Vorhersage von Proteinstrukturen durch AlphaFold.
Deepmind veröffentlichte anschließend eine wissenschaftliche Arbeit über AlphaFold inklusive des Codes und ermöglichte so unter anderem eine Open-Source-Variante der Proteinvorhersage, die aktiv in der Forschung eingesetzt wird.
Anfang 2020 sagte Deepmind mit AlphaFold dann sechs vorher unbekannte Porteinstrukturen des SARS-CoV-2-Virus vorher. Zwei dieser sechs Vorhersagen, ORF3a und ORF8, sind mittlerweile empirisch bestätigt.
AlphaFold schlägt Bestwerte und erreicht empirische Genauigkeit
Im diesjährigen CASP14-Wettbewerb trat Deepmind mit der verbesserten Vorhersage-KI AlphaFold 2.0 an. Diese erzielte entscheidende Fortschritte bei der Vorhersage von Proteinstrukturen, verkünden Deepmind und die Betreiber des Wettbewerbs gemeinsam.
„AlphaFold hat sich als fähig erwiesen, die Struktur vieler Proteine mit der Genauigkeit teurer und zeitaufwendiger Laborexperimente zu bestimmen“, schreiben die CASP-Wissenschaftler.
Hassabis spricht von Deepminds „erstem großen Durchbruch bei der Anwendung Künstlicher Intelligenz auf eine der großen Herausforderungen der Wissenschaft.“
The ultimate vision behind @DeepMind has always been to build AI and then use it to help further our knowledge about the world around us by accelerating the pace of scientific discovery. For us #AlphaFold represents an exciting first proof point of that thesis.
— Demis Hassabis (@demishassabis) November 30, 2020
AlphaFold habe von den knapp 100 im Wettbewerb zu lösenden Proteinsequenzen die Struktur von 70 ebenso präzise wie experimentelle Verfahren vorhergesagt. Das ist ein Durchbruch bei der Vorhersage der Proteinfaltung.
Alphafold 2.0 könnte die Proteinforschung um Jahre beschleunigen
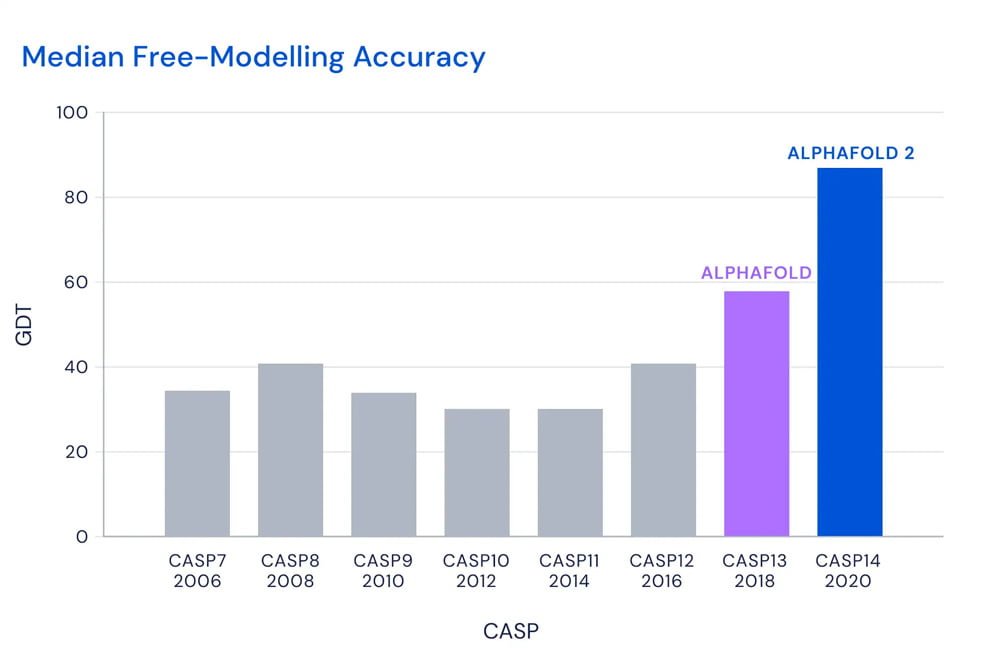
Der wichtigste Messpunkt im CASP-Wettbewerb ist der sogenannte „Global Distance Test“-Wert (GDT) mit einer Verteilung zwischen 0 und 100. Ein hoher GDT-Wert sagt aus, dass die vorhergesagten Positionen der einzelnen Aminosäurereste innerhalb des Proteins auf die Breite eines Atoms nahe an den tatsächlichen Positionen liegen. Je höher der Wert, desto genauer ist die Vorhersage.
AlphaFolds Vorhersagen lagen im Schnitt bei 92,4 GDT. Ein Wert ab 90 gilt als vergleichbar mit der Präzision experimenteller Methoden, die viele Jahre Forschung und erhebliche Ressourcen benötigen. Bei besonders herausfordernden Proteinen („Hard to Predict“) lag der Median bei etwa 87 GDT.
Hier erstaunt speziell die Geschwindigkeit, mit der die KI-Methode verbessert wurde: Im vorherigen CASP13-Wettbewerb lag der GDT-Wert noch bei etwa 53. Die besten konkurrierenden Forschergruppen erreichten dieses Jahr einen Wert von 75 GDT.
Bei einem knappen Dutzend der „Hard to Predict“-Proteine konnte AlphaFold den Forschern sogar helfen, ihre experimentellen Ergebnisse zu optimieren, berichtetet das Science Media Center.
„AlphaFolds erstaunliche Genauigkeit hat es uns ermöglicht, eine Proteinstruktur zu lösen, an der wir fast zehn Jahre festhingen und das hilft uns, unsere Bemühungen wieder aufzunehmen, zu verstehen, wie Signale über Zellmembranen übertragen werden”, sagt Professor Andrei Lupas, Direktor des Max-Planck-Instituts für Entwicklungsbiologie.
AlphaFold 2.0 setzt auf Aufmerksamkeits-Mechanismus
Möglich ist der Leistungssprung von AlphaFold 1.0 zu AlphaFold 2.0 durch eine neue KI-Architektur. Die Details sind noch nicht bekannt, da Deepminds wissenschaftliche Veröffentlichung noch aussteht. Klar ist jedoch: Die KI setzt auf den von Transformer-Architekturen bekannten Aufmerksamkeits-Mechanismus.
Dieser Mechanismus hat in den letzten Jahren die maschinelle Verarbeitung natürlicher Sprache revolutioniert, gehört nun zum Standard-Repertoire der KI-Entwicklung und ist Grundlage für die deutliche Leistungssteigerung aktueller Sprach-KIs wie Googles BERT oder OpenAIs GPT-3. Auch bei der Bildanalyse könnten Transformer der neue Standard werden und die bis dato gesetzten CNN-Netzwerke ersetzen.
Die Transformer-Architektur verarbeitet Eingabedaten im Kontext der gesamten Umgebungsdaten, etwa Wörter im Kontext eines Satzes oder Bildausschnitte in einem Gesamtbild. Vorher eingesetzte Architekturen wie LSTM-Netzwerke (“Long short-term memory”) konnten bis zu hundert Wörter an Kontext erfassen, doch die Kapazität aktueller Transformer liegt bei tausenden Wörtern und lässt sich mit zusätzlicher Rechenleistung weiter nach oben skalieren. Das erlaubt Transformer-basierten neuronalen Netzen, wesentlich komplexere Zusammenhänge zu lernen.
Der Aufmerksamkeits-Mechanismus ermöglicht AlphaFold 2.0 laut Deepmind, die Struktur eines räumlichen Graphen, der die Proteinstruktur repräsentiert, zu bilden und dabei evolutionär verwandte Sequenzen, Multiples Sequenzalignment und Repräsentationen von Aminosäurerestepaaren zu verwenden.
Die KI bezieht außerdem Informationen über physikalische und geometrische Einschränkungen für die Faltung mit ein. Das Endergebnis ist eine KI, die die finale Struktur einer Proteinsequenz direkt vorhersagt.
Das ist ein großer Unterschied zu AlphaFold 1.0: Die alte Version nutzte drei sich ergänzende neuronale Netze, um etwa die Abstände zwischen Aminosäuren vorherzusagen. Erst in einem zweiten Schritt wurde - ohne Künstliche Intelligenz - die wahrscheinlichste Proteinstruktur erzeugt.
Trainiert wurde AlphaFold mit knapp 170.000 Proteinstrukturen der Protein Data Bank und mehreren großen Datensätzen von Proteinsequenzen. Das KI-Training auf 128 von Googles speziellen KI-Kernen TPUv3 dauerte mehrere Wochen.
Großer Zuspruch aus der Wissenschaft
Von der wissenschaftlichen Gemeinschaft wird Deepminds AlphaFold 2.0 als großer Fortschritt wahrgenommen.
„Ich dachte, dass dieses Problem nicht mehr zu meinen Lebzeiten gelöst wird“, sagt Janet Thornton, Strukturbiologin am European Bioinformatics Institute in Hinxton, Großbritannien, und ehemalige CASP-Gutachterin. Sie hofft, dass der Deep-Learning-Ansatz in Zukunft die Funktion Tausender noch nicht entschlüsselten Proteinen im menschlichen Genom aufdecken kann.
Dr. Jan Kosinski vom Europäischen Laboratorium für Molekularbiologie (EMBL) in Hamburg bezeichnet AlphaFolds Erfolg als Durchbruch. Genauigkeit und Erfolgsrate seien „beispiellos“. Auch Professor Dr. Alexander Schug vom Karlsruher Institut für Technologie (KIT) nennt die bisher bekannten Ergebnisse „extrem beeindruckend“.
Doch die Wissenschaftler warnen auch vor allzu viel Übermut: Noch seien die Details zu Deepminds Methode und mögliche Schwachstellen des KI-Systems nicht bekannt. Professor Dr. Helmut Grubmüller vom Max-Planck-Institut für biophysikalische Chemie in Göttingen hält die Aussage, das Problem der Proteinfaltung sei weitgehend gelöst, für “etwas übertrieben” - schließlich liege die Trefferquote nur bei 70 von 100.
Professor Dr. Gunnar Schröder vom Forschungszentrum Jülich GmbH (FZJ) spricht von “erstaunlichen Ergebnissen”, warnt aber vor der Blackbox Künstlicher Intelligenz: „AlphaFold ist im Grunde wie eine Person, die innerhalb weniger Stunden eine Proteinstruktur experimentell bestimmen kann, uns aber nicht sagt, wie sie das macht.“
Hinzu kommt: Die von AlphaFold gelöste Proteinfaltung ist nur ein Aspekt der komplexen Interaktion zwischen Protein und anderen Molekülen oder Proteinen, die essenziell für ein Verständnis der Funktionen von Proteinkomplexen sind.
Trotz dieser Einschränkungen ist AlphaFold 2.0 der bislang größte Echtwelt-Erfolg unter Deepminds zahlreichen KI-Experimenten. Er könnte sich als einer der größten wissenschaftlichen Erfolge Künstlicher Intelligenz erweisen und als Demonstration, welche enormen Auswirkungen die Technologie auf die Wissenschaft hat.
Professor Grubmüller denkt weiter: „Mich würde sehr interessieren, ob es gelingt, diese Faltungsregeln so aus dem Deep-Learning-Algorithmus zu extrahieren, dass wir die Proteinfaltung besser vorhersagen und besser verstehen können.“
Ein solches Verständnis könnte wiederum für eine Verbesserung der Proteinfaltungsvorhersage verwendet werden. Dass sich aus Deep-Learning-Netzwerken mitunter solche Regeln extrahieren lassen, zeigten erst kürzlich KI-Forscher anhand von Problemen in der Kosmologie.
Quelle: Deepmind | Letztes Update: 1. Dezember, 7:30 Uhr